Our projects
Microbial Biodiscovery
Natural products have long been a mainstay of the pharmaceutical industry, with almost 70% of the FDA approved antibiotics either derived from, or inspired by, natural products. These figures are not surprising when you consider that natural products have evolved through millions of years of natural selection in biological systems to interact with biological systems.
The technology for pursuing natural products has matured considerably over the last three decades, making it possible to investigate the chemistry and biology of structurally novel and diverse metabolites that are present at sub-milligram levels in complex extracts. We use the latest chromatographic and spectroscopic techniques to isolate, elucidate and characterise potent and/or selective new antibiotics from a range of unique microorganisms. We assay our new antibiotics against a range of clinically important microorganisms, both in-house and in collaboration with leading national and international infectious disease researchers. The cellular targets and modes of action of these new antibiotics are also investigated (see below).
The Probio-TICK initiative

Overview
In northern Australia, ticks and buffalo fly are two of the most significant pests in cattle, costing $260 million per annum or an average $20 for every beast on every property, via direct production impacts and through the spread of tick fever, a major cause of death and illness in cattle herds. Failure to control ectoparasites is a major road-block to the improved quality, value and sustainability of the 20,000 businesses that comprise the northern Australian cattle industry. Tens of thousands more small and large businesses also depend on the vibrancy of this economic zone.
The Probio-TICK initiative is a Collaborative Research Centres Project (CRC-P) in collaboration with Microbial Screening Technologies and Meat and Livestock Australia aimed at developing a low-cost, chemical residue-free, microbial probiotic applied to the hide of cattle. By harnessing helpful microbes, we aim to provide life-long protection against cattle tick and buffalo fly.
For more information, please visit the project website at: http://www.probiotick.org/
Next-generation antibiotics

Overview
The widespread use and misuse of antibiotics, both clinically and agriculturally, has led to a dramatic increase in the prevalence of antibiotic-resistant strains of pathogenic bacteria, particularly in hospitals. New resistance mechanisms are transferring rapidly between different species of bacteria, imparting broad-spectrum antibiotic resistance to an ever-increasing microbial population. Paradoxically, the current antibiotic pipeline is almost completely dry, particularly for Gram-negative bacteria, with many drug companies downsizing or eliminating their costly antibiotic programs in favour of more profitable chronic/lifestyle illnesses. If this disturbing trend continues, deaths from previously treatable bacterial infections will very soon return to levels experienced in the early 20th century, before the dawn of the antibiotic era.
Challenges
The substantial costs associated with getting a new drug to market mean that early prioritisation of leads is essential. The most significant challenge facing modern antibiotic chemotherapy is the impact of multidrug-resistance, so a key factor in the prioritisation equation must be "mode of action". New antibiotic scaffolds that operate by the same compromised pathways as known antibiotics are clearly less attractive than those that operate by entirely new uncompromised pathways. The prioritisation of natural product antibiotics presents significant challenges as their cellular targets and modes of action are generally not known. Inevitably, these molecules end up being published as basic science or simply abandoned, squandering countless invaluable antibiotic drug leads and therapeutic opportunities.
Approach
In our group, we employ a range of techniques to rapidly link both new and historic antibiotic natural products to their cellular targets, allowing their modes of action to be determined and facilitating their prioritisation and development as chemotherapeutic agents. Some of these approaches are highlighted below:
Reverse Chemical Proteomics
Overview
The classical method of linking natural products to their cellular targets is forward chemical proteomics, whereby the natural product is either fluorescently/radioactively labelled and used to identify binding partners on a 2D protein gel, or is immobilised onto a solid support and used to "pull-down" binding partners from entire proteomes. This approach was first used in 1965 to identify a previously undescribed 57 kDa protein binding partner for the cytotoxic natural product colchicine. This protein was later shown to be the subunit of microtubules and was named "tubulin". Subsequently, forward chemical proteomics has been used to identify cellular targets for numerous other natural products, including the immunosuppressants cyclosporin and FK506, the cytotoxins didemnin B and halichondrin B, the antibiotics fumagillin and myriocin, and many others.
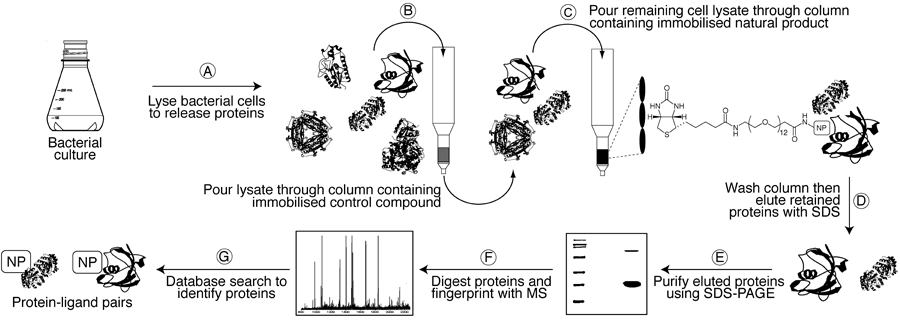
However, protein concentrations in a cell typically vary over several orders of magnitude, so it is common to isolate the most abundant rather than the most avid binding partners when using forward chemical proteomics. This can be problematic for proteins that are expressed in extremely low concentrations at any given time in a cell. Reverse chemical proteomics addresses this problem by creating a physical link between each protein and its encoding gene. Using this so-called "genotype–phenotype link", low-copy-number proteins that are recovered using a natural product affinity support can be replicated using the attached gene and subjected to subsequent pull-down experiments. This iterative approach results in amplification of weak signals (low copy-number proteins) and therefore allows the most avid binding partner to be identified.
Phage Display
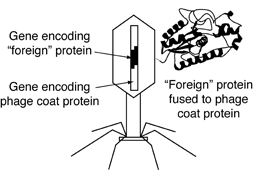
The most common method of creating a genotype–phenotype link is phage display, whereby foreign DNA is inserted into the gene that encodes the phage coat protein. When such a phage infects its bacterial host and reproduces, the protein encoded by the foreign DNA is fused to the phage coat protein, resulting in each phage particle "displaying" a different foreign protein on its surface. In most cases, the fusion of proteins does not destroy the infectivity of the phages, and the foreign proteins behave as if they were free in solution.
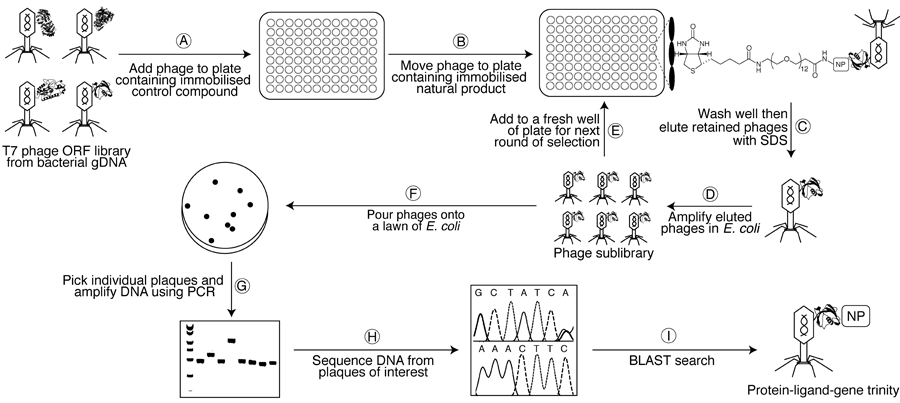
(A) A susceptible bacterial gDNA T7 phage display library is added to one well of a streptavidin-coated microtitre plate containing an immobilised biotinylated control compound and is left to incubate for 1 h to remove non-specific binders. (B) The lysate is then transferred to a second plate containing the immobilised biotinylated natural product and left to incubate for 1 h. (C–D) After washing with buffer to remove weakly-bound phages, tightly-bound phages are eluted with 1% SDS and transfected into E. coli to generate an amplified sublibrary enriched in binding partners. (E) This sublibrary is then be subjected to a second round of selection with the natural product affinity probe, and so on. (F) After 4–6 rounds of selection, the final sublibrary is poured onto a lawn of E. coli, allowing individual phage particles (plaques) to be picked. (G–I) Finally, DNA from these plaques is amplified (PCR) and sequenced to determine the identity of the displayed proteins. Human cDNA T7 phage display libraries can also be probed in an analogous manner to identify any off-target interactions.
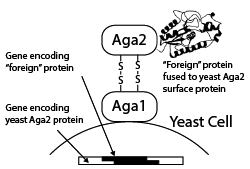
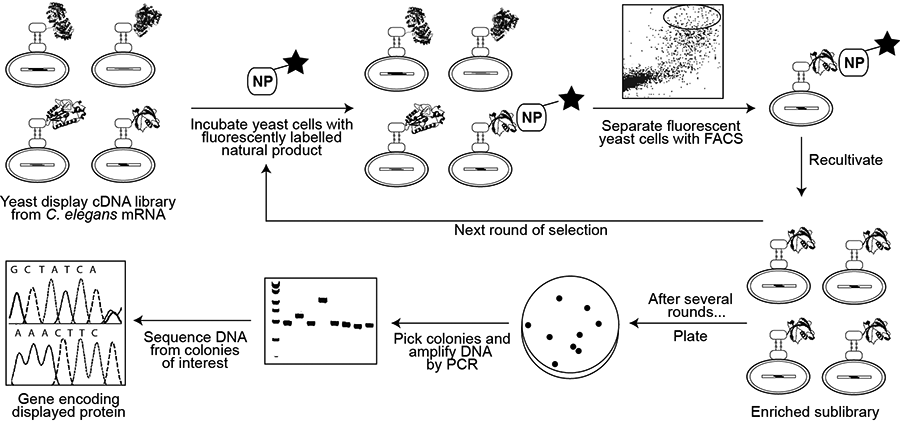
Yeast Display
A more recent alternative to phage display for creating a genotype–phenotype link is yeast display, whereby foreign proteins are displayed as fusions with the Aga2p surface protein of Saccharomyces cerevisiae and fluorescence-activated cell sorting (FACS) is used to separate yeast cells displaying proteins capable of binding to a fluorescently tagged molecule of interest. Yeast display vectors offer several advantages over phage display vectors, including the ability to post-translationally modify mammalian proteins. The use of FACS also allows for precise control over the affinity selection parameters.