SWATH-MS
A new approach to accurate quantitative whole proteome analysis. (SWATH-MS now available at APAF)
Introduction
Whole proteome analysis has traditionally been performed using data-dependent “Discovery proteomics”. While an important identification and discovery technique from a quantitative perspective it lacks reproducible selection of peptides to ensure sample-to-sample quantitation. Alternative approaches for quantitation that have been widely used are chemical tagging methods (e.g. iTRAQ or TMT) or metabolic labelling (SILAC), as these allow reliable relative quantitation between samples. Multiple reaction monitoring (MRM) MS is the gold standard for quantitation as it is highly selective and sensitive and compatible with use of isotope labelled standards. However, MRM requires prior knowledge of the analytes to be measured and acquisition methods can be laborious to develop.
Recently, with the development of faster acquisition mass spectrometers a new approach that combines discovery proteomics with reproducible quantitation has been developed. This new technique is now deployed at APAF and referred to as “SWATH-MS”. This new technique does not rely on prior knowledge of the precursor peptide ions and acquires information in a Data Independent manner. With the use of spectral libraries, SWATH-MS can be deployed to reproducibly measure the same peptides across all samples, eliminating quantitation “gaps” often seen from sample-to-sample in traditional proteomic experiments.
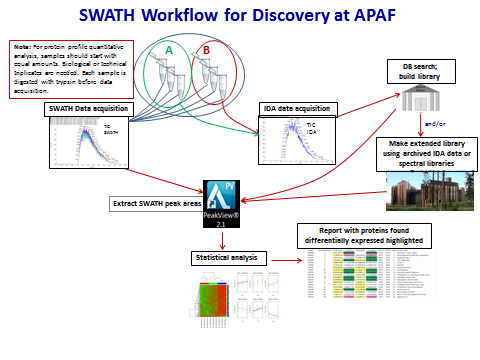
APAFs workflow
APAFs workflow for SWATH-MS involves several steps as shown in the Figure. Step 1 involves generation of a spectral library which is necessary to identify peptides acquired in SWATH-MS runs. The simplest approach for spectral library generation is conventional LC-MS/MS using samples that are representative of those to be used for SWATH-MS. Step 2 involves acquiring SWATH-MS data for each sample under analysis. Typical conditions for SWATH involve sampling the m/z 400-1200 range in 32 steps at widths of 25 m/z. This ensures comprehensive sample coverage. Step 3 involves matching the individual SWATH-MS runs against the spectral library. Currently, this matching approach is the only way to identify peptides from SWATH spectra which were acquired in a data-independent manner. It is important to note that if a peptide is not contained in the spectral library, then it will not be identified in the SWATH-MS analysis. Step 4 involves extraction of specific peptide ions to enable area-under-the-curve quantitation between samples. Once the data is extracted more sophisticated statistical analyses can be undertaken to identify changing proteins.